新火种
2024-11-17
新火种
2024-11-17 


百万级原子模拟,从头算精度,北京科学智能研究院提出AI+大尺度电子结构模拟新方法

编辑 | KX
在计算材料科学领域,准确高效地模拟材料的电子结构一直是一个非常关键而又极具挑战性的问题。基于密度泛函理论的第一性原理计算方法的高计算需求依然是大尺寸长时间材料模拟所面临的难题。
北京科学智能研究院 (AI for Science Institute, Beijing) 提出了一种基于深度学习的高效紧束缚方法,称为 DeePTB,从而高效地表示具有从头算精度的材料电子结构,极大地简化了计算复杂度,并实现百万级大尺寸结构的电子、光电响应性质的计算模拟。
当与分子动力学相结合时,DeePTB 可以同时促进原子和电子行为的有效和准确的有限温度模拟。DeePTB 的可用性弥合了电子模拟中准确性和可扩展性之间的差距,通过实现大规模电子结构计算,将推动材料科学和相关领域的发展。
相关研究以「Deep learning tight-binding approach for large-scale electronic simulations at finite temperatures with ab initio accuracy」为题,于 8 月 8 日发表在《Nature Communications》上。

虽然基于 DFT 的第一性原理方法提供了准确且通用的模拟材料电子性质的方法,但是随着系统中的原子数量增加,第一性原理的计算量急剧增加,而在真实材料或者器件体系中往往包含成百万千万量级的原子数,难以很直接使用第一性原理软件完成计算模拟。
一些复杂材料场景远远超过了 DFT 方法的模拟尺寸,一方面是因为 DFT 的自洽迭代过程复杂,另一方面,DFT 需要足够大的基组来保证精度,导致产生的哈密顿量的尺寸较大,难以进行后续的性质计算。
因此使用更小和更稀疏的矩阵来描述电子哈密顿量的紧束缚(Tight-Binding, TB)方法提供了一种更为实用的替代方案。然而,传统的 TB 方法也存在精度与效率的矛盾。例如基于 Wannier 函数的 TB 方法虽然具有较高的精度,但是其构造过程需要 DFT 自洽迭代计算,仍然限制了其在大尺寸体系下的应用。
因此发展通用的 TB 哈密顿量模型方法软件框架 DeePTB 实现精度效率以及迁移率的统一,是具有重要意义的课题。
DeePTB 方法框架及特点DeePTB 方法的整体框架图下图所示,对于给定构型中指定近邻范围内的成键原子对, 首先训练其基于物理约束下的经验公式系数,然后在此基础上提取成键原子对的局域化学环境,通过 embedding 网络构造 symmetry-preserving 描述子,并基于 fitting 网络映射为局域环境依赖的 TB 参数,突破传统模型的双中心近似,并基于 PyTorch 机器学习框架,利用 DFT 电子本征值构造损失函数,系统地进行高效的参数自动拟合。
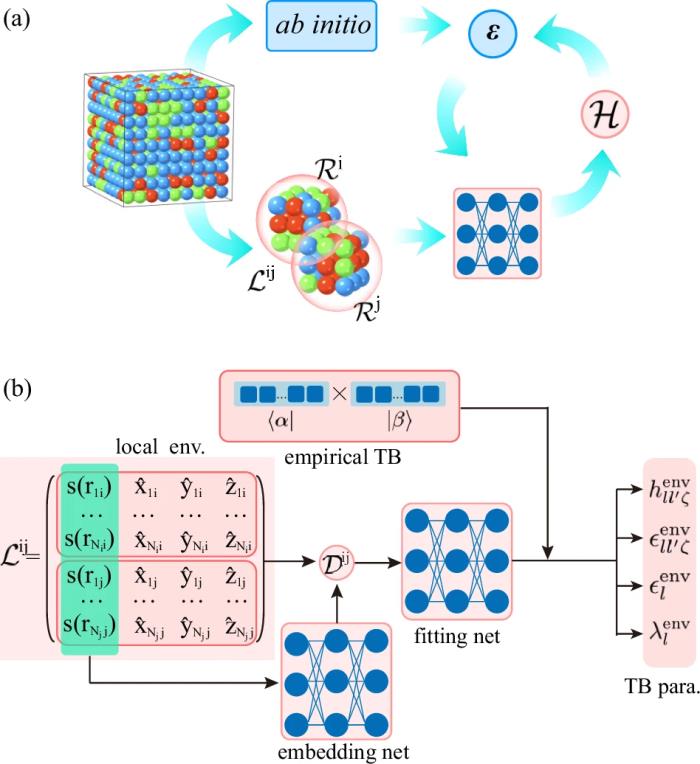
DeePTB 方法具有如下特点:
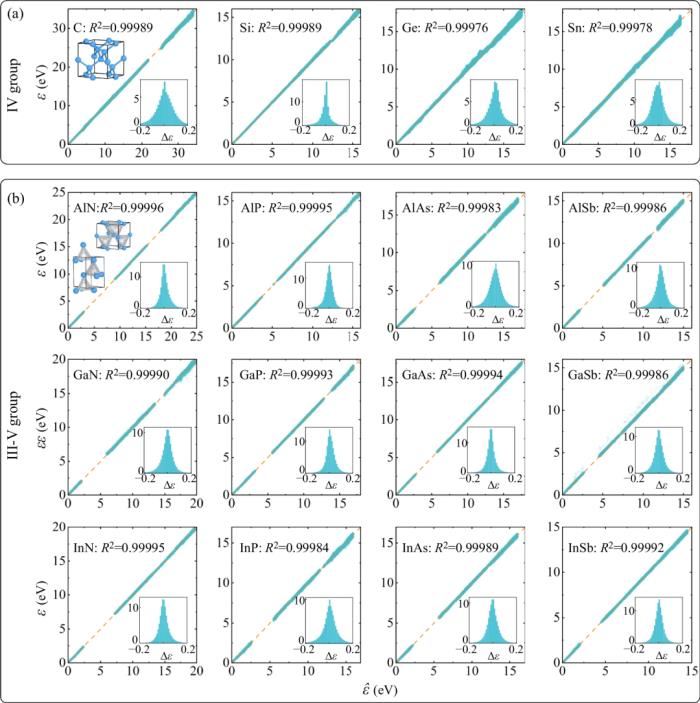
实现精度与效率的统一,通过神经网络修正实现以经验紧束缚模型计算效率并保持第一性原理的计算精度。采用 eigenvalues 作为训练标签,用户可以灵活地选择任何 DFT 软件生产训练标签,可以是平面波基组,也可以是LCAO 基组,也可以是任意泛函(LDA、GGA、even Hybrid functionals)。同时也可以轻松实现并处理自旋轨道耦合相互作用。使用更小的基组,相对完整的 LCAO DFT 哈密顿量,TB 使用的更小的基组,甚至做到只拟合费米面附近的能带。采用正交基组 TB 形式,无需额外处理交叠矩阵,因此可以接入大规模 TB 算法,例如 tight-binding propagation method (TBPM) ,轻松实现百万千万量级原子的第一性原理精度的电子性质计算。真正实现器件级尺寸的量子力学模拟。基于 Slater-Koster 框架,支持用户自定义经验 TB 拟合公式,并可以系统地增加神经网络修正,提高精度。为目前文献上存在各种经验拟合公式以及参数提供一个统一的实现和提高精度的训练平台。支持与分子动力学结合,实现有限温度下原子的动力学过程中以及结构系综采样中的电子结构和性质的模拟。预测结构扰动构型的 TB 哈密顿量以及电子结构研究人员以在电子器件中被广泛使用的 IV 族元素(C、Si、Ge、Sn)和 III-V 族化合物(如 GaAs 等)组成的半导体材料作为测试对象。
首先,进行分子动力学(MD)模拟在有限温下的结构构型采样,并基于不同 MD 轨迹的构型,使用 DFT 软件计算其对应的电子本征值作为 DeePTB 的训练和测试数据。模型测试全部体系的决定系数 (R^2≈0.9999 ) ,本征值偏差只有十几至几十个 meV 左右。其中 III-V 族化合物的测试集同时包含了立方和六方两种不同的相下的构型。
此外,DeePTB 模型还展现出了以下出色的泛化能力:
推广到更大尺寸的超胞结构,显示出极佳的尺度可扩展性。处理应变效应,准确预测应变调控下的能带结构及带隙大小。兼容不同的 DFT 基组、泛函和自旋轨道耦合效应,表现出强大的灵活性和通用性。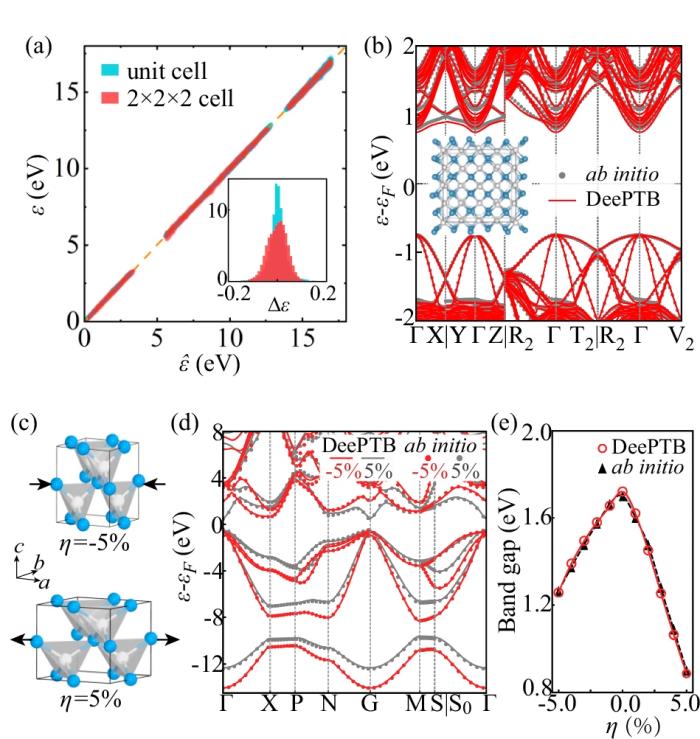
研究人员选择 III-V 族化合物 GaP 作为大尺寸建模的应用案例,构造了 50 × 50 × 50 的超胞结构。
首先基于 DP 深度势能进行 DeePMD 分子动力学模拟有限温的结构采样,然后基于得到的采样构型,利用 DeePTB 进行紧束缚模型哈密顿量的构建,并基于预测的 TB 模型使用 TBPLaS 软件实现的 TB propagation method (TBPM) 方法进行无需对角化的快速的电子性质计算,得到包括有限温下的态密度(DOS)、光电导率、介电函数以及复折射率等电子性质及光电相应,如下图所示。
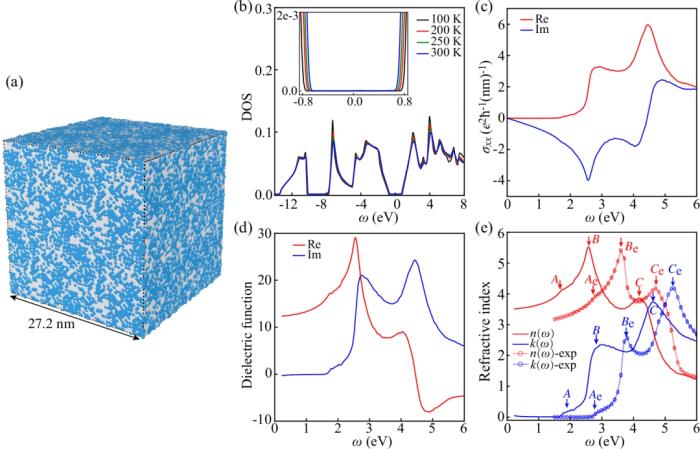
计算结果表明, DeePTB 的计算结果与文献结果符合良好,峰值位置的轻微差异主要是因为用于训练的 DeePTB 模型的交换关联泛函(GGA)倾向于低估半导体材料的电子带隙的缘故。这些结果表明了 DeePTB 高精度建模以及进行器件级尺度电子结构及性质的模拟计算的能力。
关于 DeePTB 框架的潜力对于不同的交换关联 (XC) 函数,能带结构的色散特征大致相同。因此,原则上,可以首先在计算效率高的 XC 函数(如 LDA 或 GGA)上训练模型,然后将其转移到更昂贵、更准确的函数(如 SCAN 或 HSE)。这使得能够高度准确地描述实验可观测量,以用于接近现实的材料模拟等情况下所需的大规模模拟。
此外,对于大规模样本,模拟应变对电子特性的影响是一项计算繁琐的任务。DeePTB 可以通过在较小的样本上训练模型并将其转移到更大的系统来有效地加速这些模拟。这为电子结构应变工程的理论研究带来了优势。
MD 可以提供离子自由度的模拟,这类似于晶体结构的温度探针,其中离子振动是基本现实。在需要大规模和长时间模拟的情况下,DeePTB 可用于模拟温度和结构相关的电子特性。DeePTB 使得考虑其他实际情况(如缺陷或杂质及其对电子结构的影响)成为可能和可行。
DeePTB 探索的另一个方向是模拟磁系统的特性。鉴于 DeePTB 的这些多样化潜在应用,它可以在电子模拟领域产生深远的影响。
参考内容:https://mp.weixin.qq.com/s/StetT81-UD6AGGgv-60GPA
相关推荐


- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。