

 新火种
2023-12-20
新火种
2023-12-20 


ProteinBLAST会成为过去吗?AlphaFold等对蛋白结构的搜索是否会取代对序列的搜索

编辑 | 白菜叶
像 AlphaFold 这样的蛋白质结构搜索工具会用 BLAST 取代蛋白质序列搜索吗?德累斯顿工业大学的研究团队讨论了使用结构搜索进行远程同源性检测的前景,以及为什么蛋白质 BLAST 作为领先的序列搜索工具应努力纳入结构信息。

BLAST 广泛用于分子生物学中搜索核苷酸和蛋白质序列。BLAST 推出三十年后,结构预测出现了重大突破,出现了 RoseTTAFold 和 AlphaFold 等工具。
因此,主要序列数据库中的每个蛋白质序列现在都带有一个 3D 折叠模型。虽然这不会影响(非编码)核苷酸序列,但它引出了一个问题:对 3D 蛋白质结构的搜索是否会取代对蛋白质序列的搜索。Protein BLAST 已经成为过去了吗?
虽然 BLAST 搜索是功能预测的强大工具,但它的能力是有限的。序列经过处理可以显著降解,但仍然会折叠成执行相同或相似功能的类似 3D 结构。
不同的序列,相同的结构
这种蛋白质对的例子可以在藻类和细菌的粘附分子中找到,特别是在硅藻粘附蛋白 CaTrailin_4 和细菌冰结合蛋白 FfIBP 中。该对没有可通过 BLAST 检测到的序列相似性(E 值 0.30,其中 E 值 > 0.001 不被认为是显著的)。
事实上,即使是更精细的基于序列的工具(例如 HHblits)也无法建立关系。然而,CaTrailin_4 的预测结构和 FfIBP 的已知结构非常相似,因为两者都采用由 α 螺旋持有的两个单元组成的 β 螺旋折叠 - 冰结合蛋白的拓扑特征。
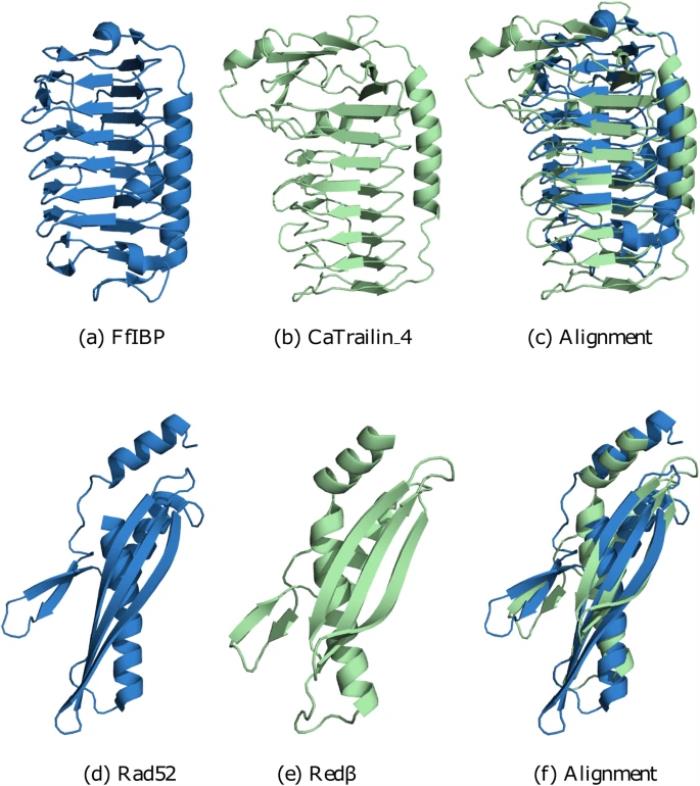
这种结构相似性可以通过所谓的模板建模分数(TM-score)来衡量,它结合了 RMSD(均方根偏差)和比对长度作为可解释的分数。大于 0.5 的 TM 分数意味着两个结构可能采用相同的折叠并且在进化上相关。CaTrailin_4 和 FfIBP 的 TM 分数为 0.6(高于 0.5 截止值)。因此,结构比较可以揭示这种惊人的相似性,而这对于 BLAST 和其他基于序列的工具(例如 HHblits)来说仍然难以捉摸。
另一个例子涉及 DNA 重组,这是复制的基本过程,其中单链退火蛋白 (SSAP) 发挥着核心作用。二十多年来,RecT/Redβ、ERF 和 RAD52 是否形成三个不同的超家族,或者只是一个超家族,一直受到怀疑和争议性的讨论。前一种观点得到了序列分析的支持,序列分析显示 RecT/Redβ、ERF 和 RAD52 之间没有明显的相似性。事实上,Rad52 和 Redβ 没有通过 BLAST 检测到的相似性(E 值 0.38)。
考虑结构会改变情况。Al-Fatlawi 团队将 RecT/Redβ、ERF 和 RAD52 的代表性结构并列在一起,结果表明,尽管缺乏序列相似性,但这些结构包含一个核心结构元件。它是寡聚反应的核心,因为它分别生成环和螺旋结构。因此,它在 RecT/Redβ、ERF 和 RAD52 中非常保守,并且可以通过结构相似性(TM 得分为 0.5)检测到,尽管缺乏任何序列相似性(见图 1 d-f)。
结构预测来拯救
这些例子表明 AlphaFold 或许能够介入 BLAST 无法发现显著相似性的领域。因此,问题出现了:如何系统地实现这一目标?为此,出现了 Foldseek、DALI 和 3D-AF-Surfer 等工具,它们分别使用自动编码器、距离矩阵对齐和专用指纹来扫描和比较结构。
虽然这些工具已经存在,但它们仍然需要更加广泛和简单,以便同序列数据库上的 BLAST 搜索竞争。需要协同作用将它们集成到经典的 BLAST 序列搜索中。最近,一项研究比较了倒数最佳 BLAST 命中和倒数最佳结构命中,并通过对序列的机器学习嵌入进行最近邻搜索,在这个方向上迈出了第一步。
为了探索这种先进工具的潜力,研究人员想要了解同一超家族的成员资格标准如何与序列和结构相似性联系起来。因此,科学家曾从 SCOPe 数据库中获得了 11,211 个具有超家族的域。这些形成 62,278,380 个结构域对,其中 225,931 个 (0.36%) 属于同一超家族,因此可以被视为同源物。
这些同源对中有多少可以分别通过序列和结构直接找到?在 E 值截止值为 0.001 时,BLAST 从 225,931 对中恢复了 16,300 对 (7%)。将界限放宽至 1,该数字增加至 25,634(11%)。但即使 E 值 < 10,也不会超过 15%。如果考虑更敏感的基于序列的方法(例如隐马尔可夫模型),这些数字会大大改善。事实上,HHblits 在最佳条件下能够检索到 175,682 对(78%),这甚至比通过结构比较(TM-score > 0.5)找到的 164,468 对(73%)要好。
然而,那 62,052,449 对不属于同一超家族的呢?在这些对中,E 值小于 0.001、1 和 10 的对分别有 0 个、9,053 个和 72,329 个。HHblits 在这 25% 中进行识别,而结构对齐的错误检测被限制在 2% 以下。HHblits 的 AUC 为 77%,结构比较为 95%,而 Blast 为 44%。较高的 AUC 分数表明,与其他超家族中的蛋白质相比,分类器能够更有效地为正确超家族中的蛋白质正确分配更高的分数。
尽管结构比较的 95% AUC 可能令人鼓舞,但高质量结构的可用性可能是一个限制。据估计,30% 的真核蛋白质含有 50 个或更多连续氨基酸的无序区域,这在 3D 结构预测中预计质量较差。这些区域适合使用 BLAST 进行序列搜索,但不适合直接结构搜索。
为了评估如此大的百分比如何扩展到整个 AlphaFold 数据库,研究人员计算了所有 AlphaFold 结构的平均置信度得分。研究人员发现 80% 的 AlphaFold 结构的 pLDDT 置信度得分为 70% 或更高,这意味着它们可以通过总体良好的主干预测进行良好建模。这意味着存在大量质量合适的结构数据。
BLAST,未来之事
BLAST 完美地满足了生物医学研究人员的许多需求,例如检测变异和密切相关的序列。然而,远程同源性检测的具体问题对于纯序列搜索来说是困难的。
在这里,结构可以比顺序更进一步。研究人员通过对数百万对结构域的演示分析来评估序列和结构相似性的这种关系。总而言之,分析表明具有严格 E 值的 BLAST 在寻找同源物方面非常精确,但并不全面。隐马尔可夫模型更敏感,但特异性有限。结构比较平衡了这两个极端。如果 BLAST 搜索包含结构数据,它可以扩展具有相似预测结构并且可能是候选同源物的命中数,而不会损害结果的质量。
如何将结构数据集成到序列搜索中尚不清楚,但一种似乎可行的方法是不直接使用结构数据,而是通过所谓的嵌入间接使用,它们是由神经网络生成的中间序列表示,构成神经网络结构预测的基础。
然而,基于嵌入和结构数据的同源检测只有以易于使用的方式提供并被社区广泛采用,才会有助于改变分子生物学。NCBI、EBI 和 Riken 等著名机构现在应该努力采用 FoldSeek 中实现的快速结构搜索,或使用嵌入来扩展经典的基于 BLAST 的蛋白质序列搜索,以便 Protein BLAST 继续成为未来的趋势。
相关推荐




- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。










