新火种
2023-12-09
新火种
2023-12-09 

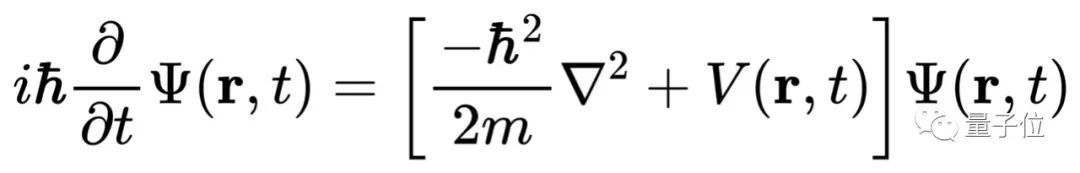

DeepMind开源薛定谔方程求解程序:从量子力学原理出发,TensorFlow实现
只要解出薛定谔方程,你就能预测分子的化学性质。但现实很骨感,迄今为止,科学家只能精确求解一个电子的氢原子,即使是只有两个电子的氦原子都无能为力。
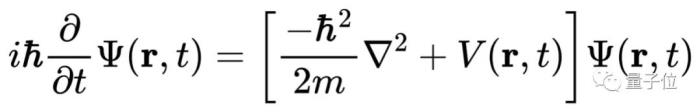
原因是有两个以上电子组成的薛定谔方程实在太复杂,根本无法精确求解。
既然找不到精确解,科学家们希望能找到一种实用的近似解求法,只要结果足够近似,也能预测原子或分子的行为。
近日,DeepMind开源了一个“费米网络”(FermiNet),用来求解分子的电子行为,在30个电子的有机分子上也达到了很高的精度。文章结果发表在期刊Physical Review Research上。
为什么叫费米网络
在量子力学中,电子没有精确的位置,我们只能从波函数预测电子在空间中出现的概率,也就是电子云。
比如氢原子的电子云就有以下几种形态。
曲面内表示电子出现的高概率区域。蓝色区域波函数为正,紫色区域波函数为负。(注:波函数平方表示电子出现的概率)
误差小于0.5%即可预测分子的能量,但这对于化学家来说远远不够,要准确预测分子的形状和化学性质,需要0.001%的精度,相当于以毫米精度测量足球场宽度。
电子在分子中不仅受到原子核的吸引力、其他电子的斥力,还遵循着量子力学中的费米-狄拉克统计:如果两个电子交换状态,波函数要反号。
这也意味着两个电子的状态不可能完全相同,否则波函数为0。这在物理中叫做“泡利不相容原理”。
费米网络正是从这个基本物理原理出发,因此DeepMind将其命名为FermiNet。
交换后符号相反,这可能会让你想到线性代数中的行列式。行列式任意两行交换,输出结果就要乘以-1。
物理学家也是这样想的,他们用所谓“Slater行列式”来表示电子波函数,但实际情况比Slater行列式要复杂得多,为了更精确表示电子行为,往往需要几百万个Slater行列式的线性组合。
工作原理
与函数线性组合相比,神经网络在表示复杂函数时往往更具有优势。
在构造FermiNet之初,研究人员就把泡利不相容原理作为第一性原理引入神经网络。
在FermiNet中,每个电子都有单独的信息流。不仅如此,他们将网络每一层所有流平均化,然后传递给下一层的每一流。这样,这些流就具有正确的反对称性要求。
而且在FermiNet行列式中的每个元素都包含所有电子,效率远远超出波函数只有单个电子的情况。
与Slater行列式不同,FermiNet是通用函数逼近器,如果神经网络层变得足够宽,则可以无限逼近真实波函数。
这意味着,如果我们正确地训练这些网络,它们应该能够将几乎完全精确的解拟合到薛定谔方程。
训练是通过最小化系统的能量来拟合FermiNet。FermiNet用蒙特卡洛方法随机选择电子构型,在每个电子排列中局部评估能量,累加每个排列的贡献,并将其最小化。
实验结果
研究人员将FermiNet用在具有10个电子以内的原子上,能量精度均在99.8%左右。
对于30个电子的环二丁烷,FermiNet算出的能量达到了97%的精度,虽然精度不是很高,但DeepMind表示,作为一种“便宜但不够准确”的方法,这是巨大的成就。
现在FermiNet已经在GitHub上开源,代码基于TensorFlow实现,如果你想用它算一算氢分子,不妨试试这串代码:
import sys
from absl import loggingfrom ferminet.utils import systemfrom ferminet import train
# Optional, for also printing training progress to STDOUTlogging.get_absl_handler().python_handler.stream = sys.stdoutlogging.set_verbosity(logging.INFO)
# Define H2 moleculemolecule = [system.Atom('H', (0, 0, -1)), system.Atom('H', (0, 0, 1))]
train.train(molecule=molecule,spins=(1, 1),batch_size=256,pretrain_config=train.PretrainConfig(iterations=100),logging_config=train.LoggingConfig(result_path='H2'
经过100次迭代后,该程序会输出一个氢原子波函数文件。官方建议最好用GPU来运行,因为他们计算乙烯分子就用8个GPU花2天时间才算出。
除了研究电子外,DeepMind还将神经网络用于其他基础科学研究,比如蛋白质折叠、玻璃态动力学、晶格量子色动力学等。
相关推荐




- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。
热门文章