新火种
2024-12-24
新火种
2024-12-24 


耗时缩短十倍以上,大规模AI方法加速原子模拟进程,推动更泛用的策略发展

编辑丨&
扩展一直是提高机器学习各个领域的模型性能和泛化的关键因素。尽管在扩展其他类型的机器学习模型方面取得了成功,但对神经网络原子间电位 (NNIP) 扩展的研究仍然有限。
该领域的主要范式是将许多物理域约束合并到模型中,例如旋转等方差等对称性约束。来自美国加州大学伯克利分校和劳伦斯伯克利国家实验室 (Berkeley Lab) 的研究团队认为,这些日益复杂的领域约束抑制了 NNIP 的扩展能力,从长远来看,这种策略可能会导致模型性能停滞不前。
为了系统地研究 NNIP 扩展属性和策略,团队提出了一种全新的专为可扩展性而设计的 NNIP 架构:高效缩放注意力原子间势 (EScAIP),意在通过注意力机制扩展模型有效提高模型表达能力。
该方法以「The Importance of Being Scalable: Improving the Speed and Accuracy of Neural Network Interatomic Potentials Across Chemical Domains」为题,于 2024 年 10 月 31 日发布于 Arxiv 预印平台。

与现有的 NNIP 模型相比,EScAIP 因使用高度优化的注意力 GPU 内核实现,效率大幅提高,推理时间至少加快 10 倍,内存使用量减少 5 倍。
团队强调,他们的方法应被视为一种哲学而非特定模型。它代表了开发通用 NNIP 的概念验证,这些 NNIP 通过扩展实现更好的表达性,并通过增加计算资源和训练数据继续有效扩展。
关于神经网络通用进化的思考
近年来,扩展模型大小、数据和计算的原理已成为提高机器学习 (ML) 性能和泛化的关键因素,涵盖从自然语言处理 (NLP)到计算机视觉 (CV)。ML 中的扩展在很大程度上取决于是否能最好地利用 GPU 计算能力。这通常涉及如何有效地将模型大小增加到较大的参数量级,以及优化模型训练和推理以实现最佳计算效率。
与这类发展并行的方向,涵盖了原子模拟,解决药物设计、催化、材料等方面的问题。其中,机器学习原子间势,尤其是神经网络原子间势 (NNIP),作为密度泛函论等计算密集型量子力学计算的替代模型而广受欢迎。
NNIP 旨在高效、准确地预测分子系统的能量和力,允许在难以用密度泛函理论直接模拟的系统上执行几何弛豫或分子动力学等下游任务。
当前的 NNIP 主要基于图神经网络 (GNN)。该领域的许多有效模型越来越多地尝试将受物理启发的约束嵌入到模型中,这些约束包括将预定义的对称性(例如旋转等方差)合并到 NN 架构中,以及使用复杂的输入特征集。
团队认为,这些日益复杂的领域约束抑制了 ML 模型的扩展能力,并且可能会随着时间的推移在模型性能方面趋于平稳。随着模型规模的增加,可以假设施加这些约束会阻碍有效表示的学习,限制模型的泛化能力,并阻碍有效的优化。其中许多功能工程方法并未针对 GPU 上的高效并行化进行优化,进一步限制了它们的可扩展性和效率。
基于这些理解,他们开发了高效缩放注意力原子电位 (EScAIP)。该模型在各种化学应用中实现了最佳性能,包括在 Open Catalyst 2020、Open Catalyst 2022、SPICE 分子和材料项目 (MPTrj) 数据集上的最佳性能。
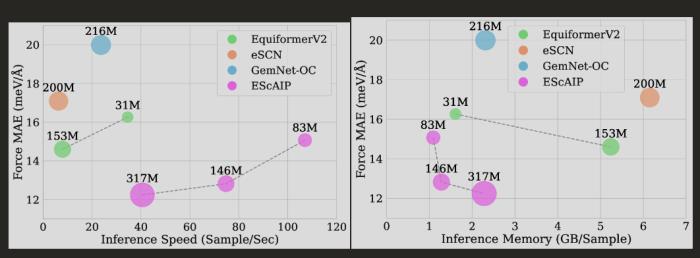
EScAIP 可以很好地与计算一起扩展,并且其设计方式将随着 GPU 计算的不断进步而进一步提高效率。
模型的种子与未来的枝芽
这些模型通常经过训练,可以根据系统属性(包括原子序数和位置)预测系统能量和每原子力。模型分为两类:基于组表示节点特征的模型,以及基于笛卡尔坐标表示的节点特征的模型。
NNIP 领域也越来越关注使用量子力学模拟生成更大的数据集,并使用它来训练模型。有一种趋势是将物理启发的约束纳入 NNIP 模型架构,例如所有将对称约束合并到模型中的组。然而,还有其他工作路线并没有试图直接在 NNIP 中构建对称性,而是尝试“近似”对称性。
通过消融研究,该团队系统地研究了缩放神经网络原子间势 (NNIP) 模型的策略。在确认了高阶对称性(旋转阶数
相关推荐


- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。