新火种
2024-11-27
新火种
2024-11-27 


探索蛋白质动态变化,新AI方法JAMUN比标准MD模拟更快、更准确

编辑 | 白菜叶
蛋白质结构的动态变化对于理解其功能和开发靶向药物治疗至关重要,尤其是对于隐蔽的结合位点。然而,现有的生成构象集合的方法存在效率低下或缺乏通用性的问题,无法在训练系统之外发挥作用。
分子动力学 (MD) 模拟是当前探索蛋白质运动的标准,但计算成本高昂,且受短时间步长要求的限制,因此难以捕捉较长时间尺度上发生的更广泛蛋白质构象变化。
Prescient Design 和Genentech的研究人员推出了 JAMUN(walk-Jump Accelerated Molecular ensembles with Universal Noise),这是一种新颖的机器学习模型,旨在通过实现对蛋白质构象集合的有效采样来克服这些挑战。
该研究以「JAMUN:Transferable Molecular Conformational Ensemble Generation with Walk-Jump Sampling」为题,于 2024 年 10 月 18 日发布在 arXiv 预印平台。

JAMUN 将 Walk-Jump Sampling(WJS)扩展到 3D 点云,代表蛋白质原子坐标。通过利用 SE(3) 等变去噪网络,JAMUN 可以以比传统 MD 方法或当前基于 ML 的方法高得多的速度对任意蛋白质的玻尔兹曼分布进行采样。
JAMUN 还表现出了显著的转移到新系统的能力,这意味着它甚至可以为不属于其训练数据集的蛋白质结构生成可靠的构象集合。
所提出的方法基于「Walk-Jump Sampling」的概念,即将噪声添加到干净的数据中,然后训练神经网络对其进行去噪,从而实现平稳的采样过程。
JAMUN 在「Walk」阶段采用朗之万动力学,这已经是分子动力学 MD 模拟中的标准方法。
然后,「Jump」步骤会投射回原始数据分布,从而将过程与每次重新开始分离,而这通常是扩散模型所为。通过分离 Walk 和 Jump 步骤,JAMUN 可以平滑数据分布,足以解决采样困难,同时保留 MD 数据固有的物理先验。
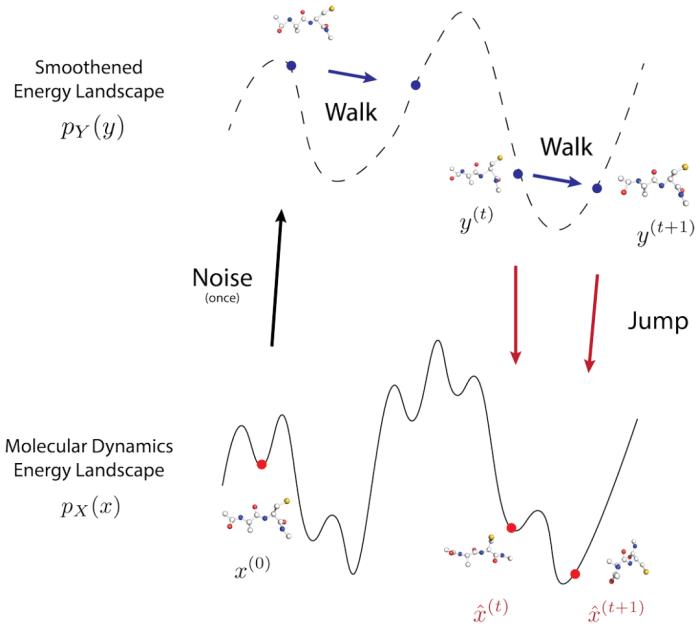
JAMUN 是在两种氨基酸肽的分子动力学模拟数据集上进行训练的,并成功推广到未见过的肽。结果表明,JAMUN 可以比标准 MD 模拟更快地对小肽的构象集合进行采样。
例如,JAMUN 在一小时内计算生成了具有挑战性的封端肽的构象状态,而传统的 MD 方法需要更长的时间来覆盖类似的分布。
JAMUN 还与可转移玻尔兹曼生成器 (TBG) 模型进行了比较,展示了显著的加速和相当的准确性,尽管它仅限于玻尔兹曼模拟而不是精确采样。
JAMUN 提供了一种强大的新方法来生成蛋白质的构象集合,平衡了效率和物理精度。它能够以比 MD 快得多的速度生成集合,同时保持可靠的采样,使其成为蛋白质结构预测和药物发现应用的有前途的工具。
未来的工作将侧重于将 JAMUN 扩展到更大的蛋白质,并改进去噪网络以实现更快的采样。
通过利用 Walk-Jump 采样,JAMUN 朝着可推广、可转移的蛋白质构象集合生成解决方案迈出了重要一步,这对于生物学理解和药物创新都至关重要。
论文链接:https://arxiv.org/abs/2410.14621
相关内容:https://www.marktechpost.com/2024/10/21/jamun-a-walk-jump-sampling-model-for-generating-ensembles-of-molecular-conformations/
相关推荐




- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。