

 新火种
2024-11-15
新火种
2024-11-15 


分类准确率达99%,山大团队提出基于对比学习的基因数据分类方法

编辑 | 萝卜皮
深度神经网络模型的快速进步显著增强了从微生物序列数据中提取特征的能力,这对于解决生物学挑战至关重要。然而,标记微生物数据的稀缺性和复杂性给监督学习方法带来了巨大的困难。
为了解决这些问题,山东大学的研究人员提出了 DNASimCLR,这是一个专为高效基因序列数据特征提取而设计的无监督框架。
DNASimCLR 利用卷积神经网络和基于对比学习的 SimCLR 框架,从不同的微生物基因序列中提取复杂特征。预训练在两个经典的大型未标记数据集上进行,包括宏基因组和病毒基因序列。后续分类任务通过使用之前获得的模型对预训练模型进行微调来执行。
DNASimCLR 的多功能性使其在涉及新基因序列或以前未见过的基因序列的场景中表现良好,使其成为基因组学中各种应用的宝贵工具。
该研究以「DNASimCLR: a contrastive learning-based deep learning approach for gene sequence data classification」为题,于 2024 年 10 月 14 日发布在《BMC Bioinformatics》。

即使是目前最全面的微生物基因数据库也存在数据和标签缺失的问题,这严重限制了许多监督式深度学习方法的有效性。解决这一不完整性是一项迫切需要关注的挑战。
本文针对微生物基因序列数据的表征学习问题,提出了一种基于对比学习的神经网络特征提取方法。
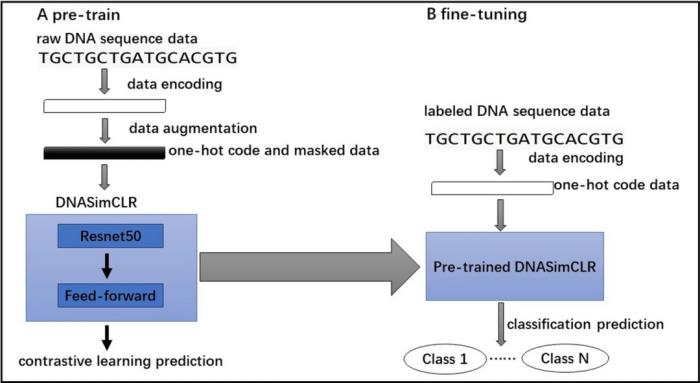
图示:DNASimCLR 框架概述。(来源:论文)
DNASimCLR 的工作流程主要包括两个阶段:对比学习的预训练阶段和分类网络的微调阶段。在预训练阶段,研究人员使用 One-Hot 编码方法将未标记的原始 DNA 基因序列数据转换为适合机器学习的格式。
在预训练阶段,对 One-Hot 编码数据进行随机掩码处理,生成训练数据集。在此阶段,研究人员采用 SimCLR 框架模型来获取未标记序列的向量表示。该过程通过对比学习将基因序列嵌入到固定维度的高维空间中。
在微调阶段,利用预训练阶段得到的特征提取模型,对标注数据采用不进行掩蔽操作的One-Hot编码方法进行编码。研究人员继续进行分类预测的训练,最终得到一个能够确定 DNA 序列类别的分类网络
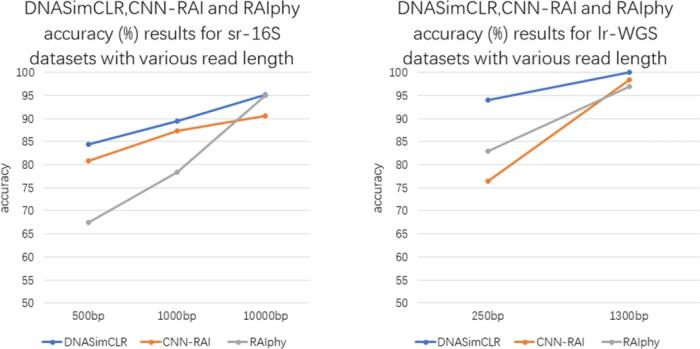
在性能评估方面,该团队对不同来源的基因组数据库进行了测试。
研究人员用 DNASimCLR 对不同长度(250 bp、500 bp、1000 bp、1300 bp 和 10,000 bp)的读段序列进行了分类和短序列病毒宿主预测,实现了 99% 的显著分类准确率,实现了显著的准确性提升。
并且,这项研究的意义是多方面的。
首先,首次将对比学习应用到微生物基因序列数据的表征学习中,发展了一种新的针对基因数据的数据处理方法,突破了传统SimCLR方法仅适用于图像数据的局限性,拓展了对比学习的应用领域。
其次,该研究提出的微生物基因序列数据分类器在性能上表现出了大幅的提升,为卷积神经网络方法在处理生物数据方面的发展开辟了新的机遇。
第三,由于预训练阶段和分类阶段的分离,该方法可以轻松应用于其他基因组学问题,例如蛋白质功能预测和新病毒检测。
总之,DNASimCLR 代表了利用自监督学习模型进行微生物基因序列特征提取的先进探索。这种方法有可能在生物信息学领域引入创新概念,提供通过卷积神经网络获取生物序列特征的途径。
论文链接:
相关推荐




- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。










