

 新火种
2024-11-15
新火种
2024-11-15 


不懂AI、不会编码?如何轻松拿捏AlphaFold准确预测蛋白结构

编辑 | 萝卜皮
自 2021 年公开发布以来,用 AlphaFold2 (AF2) 预测蛋白质结构研究生物学问题,已经成为一种常见做法。
ColabFold-AF2 是 Google Colaboratory 内部的开源Jupyter Notebook,也是一个命令行工具,可让你轻松使用 AF2,同时展示了其高级选项。
ColabFold-AF2 优化了 AF2 模型的使用,缩短了实验的周转时间。
在这里,韩国首尔国立大学的研究团队发布了一项 protocol,可以帮助研究人员更方便简洁的使用 AF2,同时介绍了一些操作技巧。
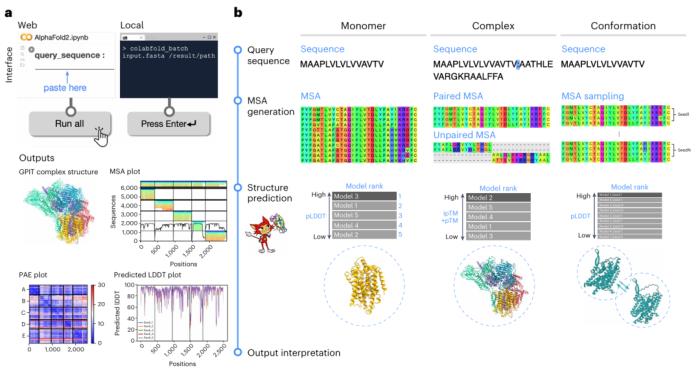
在此protocol中,研究人员通过以下三种场景引导读者了解 ColabFold 的最佳实践:(i)单体预测,(ii)复合物预测和(iii)构象采样。
前两种情况涵盖了经典的静态结构预测,并在人类糖基磷脂酰肌醇转酰胺酶蛋白上进行了演示。第三种场景通过预测人类丙氨酸丝氨酸转运蛋白 2 的两种构象展示了 AF2 模型的另一种用例。
用户可以通过 Google Colaboratory 运行该 protocol,而无需计算专业知识,高级用户也可以在命令行环境中运行该 protocol。
该研究以「Easy and accurate protein structure prediction using ColabFold」为题,于 2024 年 10 月 14 日发布在《Nature Protocols》。

关于 AlphaFold
仅从蛋白质序列预测其三维结构长期以来一直是结构生物学领域的一项艰巨任务。机器学习模型的进步在实现这一目标方面取得了重大进展。
AlphaFold2 (AF2) 和RoseTTAFold代表了这些突破性的模型。它们首次提供了计算方法,能够在提供足够的序列信息的情况下生成与实验解决的结构几乎无法区分的蛋白质结构预测。具体来说,AF2 是一个端到端神经网络,由两个主要模块组成。
第一个模块处理有关输入氨基酸 (AA) 序列 (查询) 的信息,并根据多序列比对 (MSA) 生成关于哪些 AA 相互接触的假设。第二个模块汇总这些假设以预测结构(即每个原子的 3D 坐标)。
AF2 网络背后的两个关键思想是使用深度学习注意机制,这使网络能够更好地识别接触的 AA,并通过多次通过模块来细化预测。类似的原则指导了 RoseTTAFold 的设计,从而产生了一种不同的网络架构,其卓越准确性可与 AF2 相媲美。
AF2 最初设计用于预测单链(单个蛋白质链)的结构。然而,其模型已成功用于预测多链或复合物之间的相互作用。
此外,AF2 模型得到了进一步开发和训练,专门针对多聚体输入,从而产生了 AlphaFold-multimer。从那时起,已经开发了许多其他使用 AF2 或其概念作为基础的模型。
关于 ColabFold
ColabFold 是一个集成的蛋白质预测解决方案,旨在简化用户的结构建模过程。因此,它既提供了各种蛋白质预测模型的简单界面,也提供了预处理和后处理程序。ColabFold 有两个界面:基于 Web 的界面(使用 Google Colaboratory notebooks,以下简称 Colab)和命令行工具。
其基于 Web 的界面包括五个 notebooks:AlphaFold2.ipynb (用于使用 AF2 或 AF2-multimer)、RoseTTAFold2.ipynb、RoseTTAFold.ipynb(主要用于旧版)、ESMFold.ipynb 和 OmegaFold.ipynb(在下面的 ColabFold-AF2 替代品中简要介绍)。基于 Web 的界面需要免费注册 Colab,主要用于进行单个或小批量预测。
命令行界面仅包含 AF2 和 AF2-multimer 预测模型,并允许通过处理多个输入序列进行批量预测。由于 AF2 模型是目前发布的最准确的模型之一,因此 protocol 专注于 AlphaFold2.ipynb 和命令行界面,它们统称为「ColabFold-AF2」。
由于其简单性和功能性,ColabFold 已被广泛用于众多研究,其公共 MSA 服务器每天被使用数万次。它的适用性涵盖了许多生物学领域。该论文将指导读者如何使用 ColabFold-AF2 解决类似的生物学问题。
新的 protocol
在 protocol 中,首尔国立大学的研究人员修改并扩展了 del Alamo 等人提出的 protocol,并通过使用人类丙氨酸丝氨酸转运蛋白 2(ASCT2,一种 Na+ 独立的中性 AA 转运蛋白)来展示这种能力。
该转运蛋白是一种同型三聚体,至少有两种构象,具体取决于它是面向细胞外(向外)还是细胞内(向内)。
通过调整 ColabFold-AF2,该团队展示了预测这些不同结构状态的能力。
论文概述了使用 ColabFold-AF2 进行单体(程序 1、2)和复合物(程序 3、4)预测以及构象采样(程序 5、6)的临时方法的综合方案。
程序 1、3、5 是使用 Colab 的基于 Web 的方法,程序 2、4 、6 是使用命令行界面的本地方法。

ColabFold-AF2 允许通过单击进行蛋白质预测,称为「快速启动」。要快速启动,请打开 Web 浏览器并导航到 https://alphafold.colabfold.com。接下来,将目标蛋白质的 AA 序列粘贴到 query_sequence 字段中,然后单击菜单栏中的 Runtime → Run all(下图)。
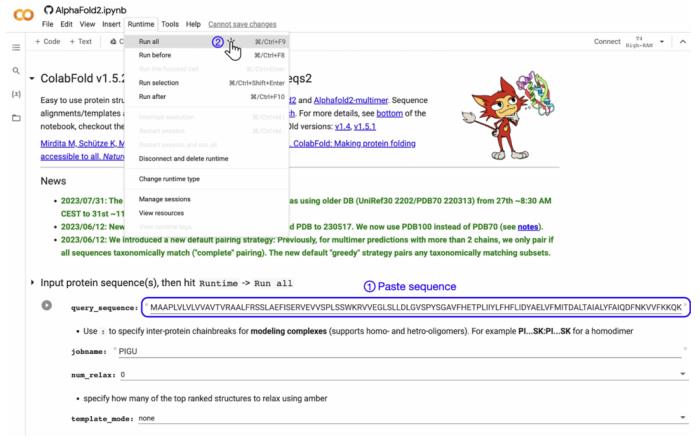
值得注意的是,如果连续运行多个预测,请单击「重新启动会话并运行全部」而不是「运行全部」,以避免任何冲突或内存泄漏。这将连续执行 Notebook 中的每个单元而不间断,并且当前正在运行的单元由左侧的旋转圆圈表示。默认情况下,ColabFold 会计算五个结构模型。
生成每个模型后,会显示预测的结构和结果图。处理完所有单元格后,将出现一个弹出窗口,提示你下载各种结果文件作为单个压缩 (zip) 文件。请注意,这些步骤的运行时间可能因 Colab 分配的 GPU 而异。
使用 protocol 的命令行版本需要对 Unix/Linux shell有基本的了解,并且需要能够处理 AF2 模型的工作站。
研究人员使用人类 GPIT 蛋白演示程序 1-4。程序 1 和 2 将其五个亚基 PIGU、PIGK、PIGT、PIGS 和 GPAA1 中的每一个预测为单体,程序 3 和 4 将它们联合预测为复合物。在程序 5 和 6 中,他们使用人类 ASCT2。
为了区分方便,研究人员根据参数的作用将其分为几类。但是,这些参数分布在 Notebook 的多个单元格中。在每个过程中,论文详细介绍了用户如何向 ColabFold 提供输入蛋白质序列,BOX3 提供了有关 ColabFold 可接受的输入和输出格式的完整详细信息。

BOX4 提供了 ColabFold-AF2 计算的各种置信度测量的信息,BOX5 适用于不想使用默认 ColabFold 服务器进行 MSA 计算的用户。


此 protocol 的预期结果部分包含有关解释 ColabFold 图表和输出的一般说明,然后演示了每个程序示例的解释过程。此 protocol 主要面向旨在进行结构分析的生物学家,不需要编码专业知识。
详细信息请阅读原论文。
论文链接:https://www.nature.com/articles/s41596-024-01060-5
相关推荐


- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。










