

 新火种
2023-12-20
新火种
2023-12-20 


GPU上运行速度比现有模型快3-7倍,IU团队使用全卷积神经网络进行准确的从头肽测序

编辑 | 萝卜皮
从头肽测序不依赖于全面的靶序列数据库,这为科学家提供了一种从串联质谱中识别新肽的方法。然而,当前的从头测序算法的准确性和覆盖率较低,这阻碍了它们在蛋白质组学中的应用。
印第安纳大学(Indiana University,IU)的研究人员提出了 PepNet,一种用于高精度从头肽测序的全卷积神经网络。PepNet 将 MS/MS 谱(表示为高维向量)作为输入,并输出最佳肽序列及其置信度得分。PepNet 模型使用来自多个人类肽谱库的总共 300 万个高能碰撞解离 MS/MS 谱图进行训练。
评估结果表明,PepNet 在肽级精度和位置级精度方面均显著优于当前性能最佳的从头测序算法(例如 PointNovo 和 DeepNovo)。PepNet 可以对数据库搜索引擎未识别的大部分光谱进行测序,因此可以用作数据库搜索引擎的补充工具,用于蛋白质组学中的肽识别。
此外,PepNet 在 GPU 上的运行速度分别比 PointNovo 和 DeepNovo 快 3 倍和 7 倍,因此更适合大规模蛋白质组数据的分析。
该研究以「Accurate de novo peptide sequencing using fully convolutional neural networks」为题,于 2023 年 12 月 2 日发布在《Nature Communications》。

过去十年见证了质谱技术的巨大进步,特别是液相色谱耦合串联质谱(LC-MS/MS)。随着通量和灵敏度的提高,LC-MS/MS 已成为对包括人类在内的高等生物体的各种生理(例如疾病)条件下的整个蛋白质组规模的蛋白质功能研究最广泛使用的方法之一。
在典型的蛋白质组学实验中,获得 MS/MS 谱图后,第一步(可以说是最重要的步骤)是从这些谱图中识别肽。人们开发了许多算法来解决这个问题,这些算法主要分为三类:蛋白质数据库搜索、光谱库搜索和从头测序。
蛋白质数据库搜索是肽鉴定的主要方法。肽序列标签法和 Sequest 算法是该类较早的算法。最近的开发包括 Mascot、X!Tandem、OMSSA、MyriMatch、Protein Prospector 和 MSGF+。这些方法将实验光谱与蛋白质数据库中肽生成的理论光谱进行比较,并报告那些可能真实的肽光谱匹配(PSM)。
相比之下,光谱库搜索方法将新采集的 MS/MS 光谱与包含早期计算分析中使用的先前表征的实验光谱的库进行比较。由于 MS/MS 数据的重复性和再现性的提高以及大量实验光谱的可用性不断提高(例如,来自蛋白质组学数据存储库和大规模合成肽项目),光谱库搜索方法已被越来越多地采用,并在 X!hunter、SpectraST 和 msSLASH 等软件工具中实施。
最后,从头测序算法尝试直接从 MS/MS 谱图中导出肽序列,而不使用谱库或蛋白质序列数据库等参考资料。许多从头测序算法采用图论公式,通过动态规划算法和自适应评分方案来计算谱图中的最长路径。随着高分辨率 MS 仪器的进步,从头测序算法的性能显著提高,特别是在更复杂的评分方案中。
近期,DeepNovo 及其后继模型 PointNovo 是使用深度学习算法开发的,该算法可以从大量肽的 MS/MS 谱图中自动学习与肽序列相关的碎片离子模式,并报告了性能的改进。这些方法利用深度神经网络 (DNN) 架构来捕获输入串联质谱中碎片离子之间的依赖性,随后将其用于按顺序构建肽。
尽管这些方法比传统的从头测序算法表现出更好的性能,但研究人员观察到它们可以对相对较少的长肽进行测序,特别是来自 Charge 3+ MS/MS 谱图,这可能是由于对碎片离子之间复杂的长程模式进行建模的挑战。另一方面,PredFull 采用的卷积神经网络(CNN)架构进行完整的 MS/MS 谱预测,展示了 CNN 学习 MS/MS 谱中复杂模式的优势。
因此,IU 的研究人员开发了一种名为 PepNet 的深度学习模型,与之前的方法相比,该模型显著提高了串联质谱从头肽测序的性能。
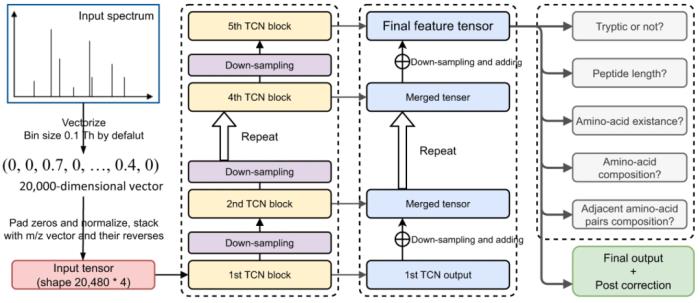
图:PepNet 的神经网络架构。(来源:论文)
研究人员首先证明 PepNet 能够对人类 MS/MS 谱图进行高精度测序,然后证明 PepNet 可以在来自许多非人类生物体的 MS/MS 数据中始终表现良好。此外,对未识别光谱的从头测序结果表明,PepNet 有能力从 MaxQuant 忽略的光谱中发现大量识别,产生的识别比 MaxQuant 之前检测到的识别多几倍。
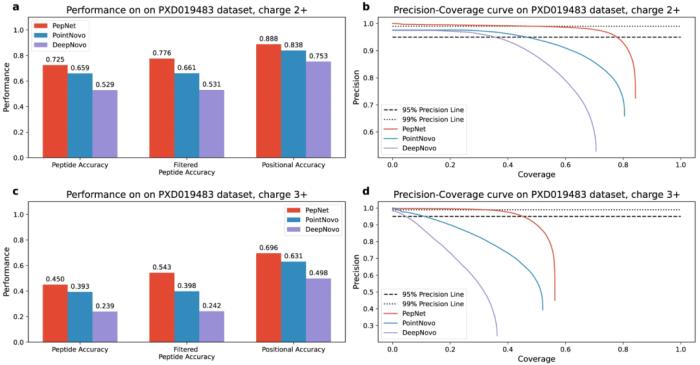
图:PepNet、PointNovo 和 DeepNovo 在人类蛋白质组数据集中的 2+ 和 3+ 电荷谱上的准确度和精度覆盖曲线。(来源:论文)
平均而言,在相当的精度水平下,PepNet 可以比其他工具多 2.5-19 倍地测序未识别的光谱。
这表明,尽管 PepNet 是使用 MaxQuant 等数据库搜索工具测序的肽进行训练的,但 PepNet(和其他 de novo 算法)不受训练样本的特定肽知识的限制。因此,PepNet 可以用作蛋白质组数据分析的强大工具,特别是当没有全面的目标蛋白质序列数据库时(例如,在宏蛋白质组学中)。
研究人员认为,高精度肽测序的能力将使从头肽测序在蛋白质组学数据分析中的应用不断增加。除了论文里介绍的 HCD 谱图肽测序之外,PepNet 还可以扩展到使用其他碎片方法获取的 MS/MS 谱图,例如电子转移解离(ETD)、电子转移/高能碰撞解离(EThcD)、光解离(PD)和红外多光子解离(IRMPD)。这些方法通常被认为会产生复杂的 MS/MS 谱图,其中嵌入复杂 MS/MS 谱图的丰富信息有望提高从头肽测序的准确性。
总而言之,PepNet 显著提高了从头肽测序的准确性,因此可以作为数据库搜索引擎的补充工具,用于蛋白质组学中的肽鉴定。因此,研究人员认为 PepNet 将提高蛋白质组学数据分析的效率,并造福生命科学研究。
相关推荐



- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。










