

 新火种
2024-06-11
新火种
2024-06-11 


清华系细胞大模型登Nature子刊!能对人类2万基因同时建模,代码已开源
生命科学领域的基础大模型来了!
来自清华、百图生科的团队提出的单细胞基础大模型scFoundation,登上Nature Methods。

该模型基于5000万人类单细胞测序的数据进行训练,拥有1亿参数,能够同时处理约20000个基因。
团队在模型架构上进行了创新,相同参数量下计算时间是传统Transformer架构的3%左右。相关研究成果也被NeurIPS2024接收。
清华大学自动化系博士研究生郝敏升为该论文的第一作者。清华大学张学工教授,马剑竹教授,百图生科宋乐教授为通讯作者。
作为基础模型,它在细胞测序深度增强、细胞药物响应预测和细胞扰动预测等下游任务中表现出卓越的性能提升,并为基因网络推断和转录因子识别提供了新的研究思路。
细胞基础大模型登Nature子刊
通过在大规模语料库上的训练,大模型才具备了基本的语言理解和识别能力。
在生命科学领域,细胞可以被视为拥有自身“语言”的基本结构和功能单元,由DNA序列、蛋白质和基因表达值等构成无数“词语”的“句子”。
那么随之而来的问题是:
目前训练大规模单细胞数据主要存在以下三点挑战:
1、基因表达预训练数据需要涵盖不同状态和类型的细胞景观。然而目前大多数单细胞数据组织松散,全面完整的数据库仍然缺失。
2、在训练过程中,传统的transformer难以处理近20000个蛋白质编码基因构成的“句子”,这使得现有工作通常不得不将模型限制在一小部分预选的基因列表上。
3、 不同技术和实验室的单细胞转录数据在测序深度上存在差异,这妨碍了模型学习统一且有意义的细胞和基因表示。
针对这些问题,研究团队首先收集了超过5000万个涵盖各个器官、肿瘤和非肿瘤的大规模人类单细胞数据集用于训练。
与大型语言模型中的“词-向量”转换不同,scFoundation通过巧妙设计,将连续的基因表达值转化为向量。
针对单细胞数据的高稀疏性以及零值和非零值所包含信息量的差异,研究团队设计了一个非对称编码模块。
该模块在保持相同参数规模的情况下,所需的计算量仅为传统语言模型Transformer的3.4%。
此外,研究团队还提出了一种测序深度感知的预训练任务“read-depth-aware (RDA)”,能够对测序深度进行降采样,使得模型在预训练阶段在完成传统的掩膜恢复任务外,还能够由低质量细胞恢复高质量细胞的基因表达信息。
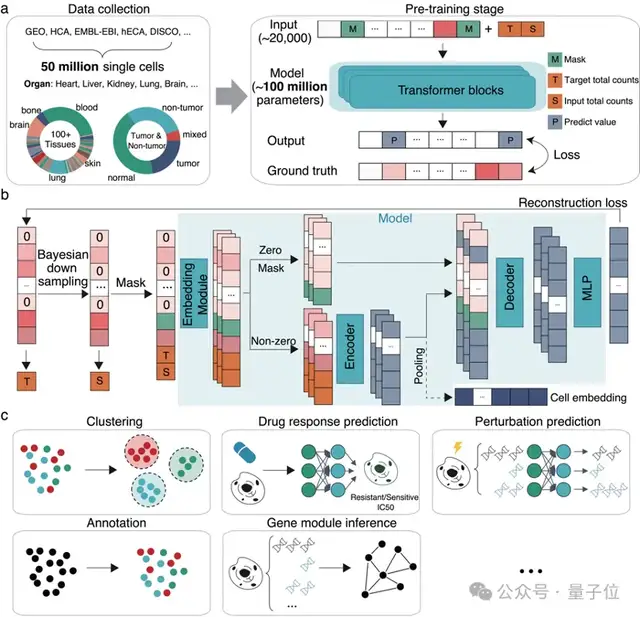
△scFoundation模型及下游应用场景
两种应用范式
scFoundation的应用范式主要包括开箱即用和微调两种:
从scFoundation得到表征,进一步利用下游方法分析。训练scFoundation一层和针对各个任务的MLP头,进行标签预测。
在开箱即用范式上,受益于RDA预训练任务,将scFoundation应用于细胞测序深度增强任务,在不需要进一步微调的情况下达到了比现有训练方法相当甚至更好的效果。
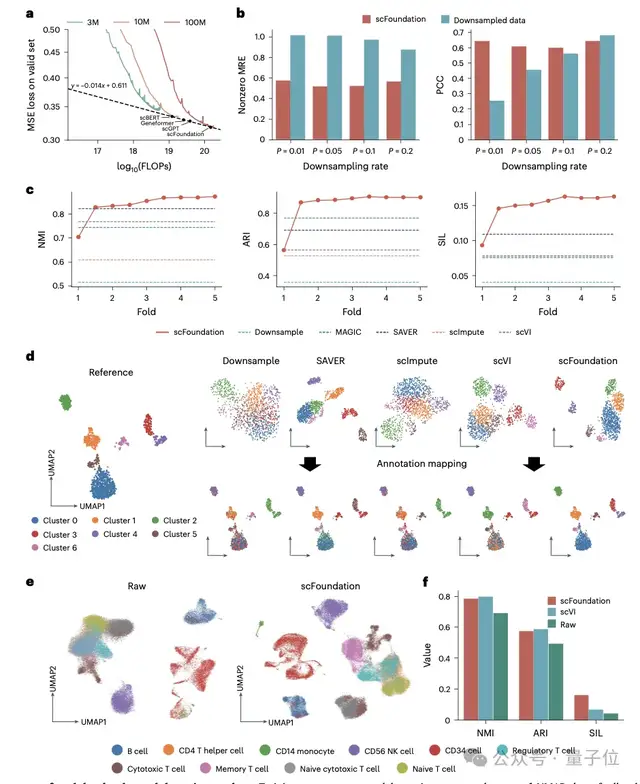
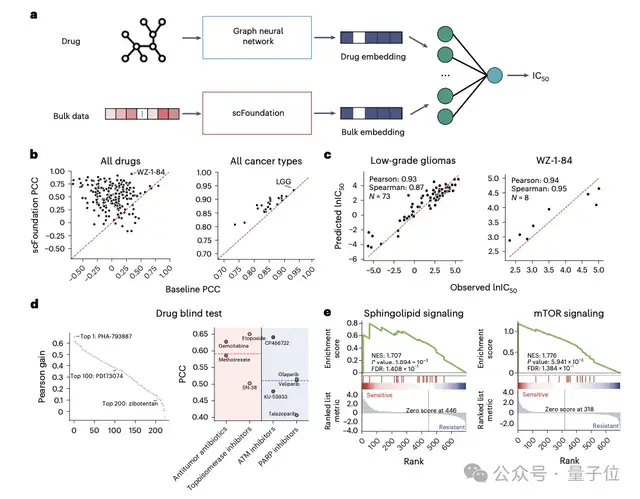
此外,通过构建模型预测细胞对癌症药物干预的反应,对指导抗癌药物的设计及理解癌症的生物学机制至关重要。
基于scFoundation提取的Bulk基因表达数据,能够预测药物半最大抑制浓度IC50及单细胞水平的药物敏感性,显示出在几乎所有药物和癌症类型上预测效果均有显著提升。
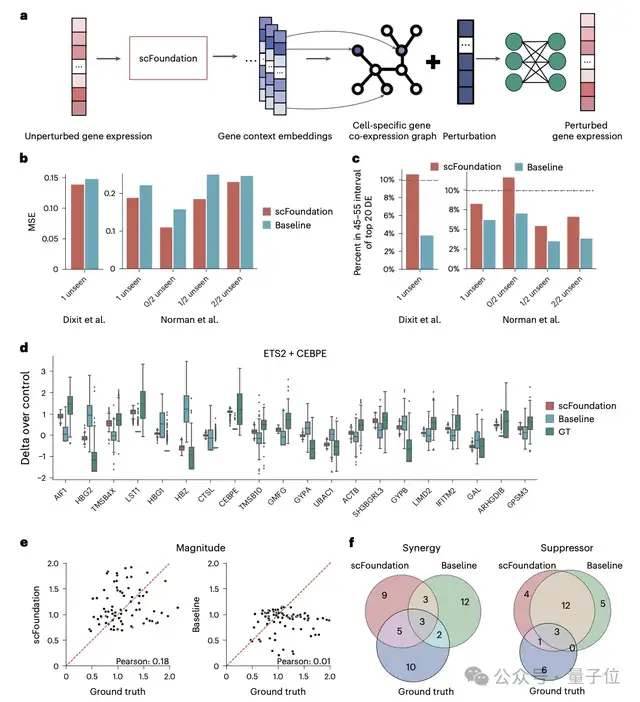
而在细胞扰动预测任务中,通过提取单个细胞的基因表征来构建特定的基因共表达网络,scFoundation成功捕捉了不同条件下的细胞和基因表征,显著提高了单/双扰动预测的准确度。
此外,基因表征还可用于构建针对特定细胞类型的基因网络。研究团队在T、B和Monocyte细胞类型中识别出了特异的基因模块和转录因子。在微调应用方面,scFoundation在细胞类型标注任务中的效果远超传统方法。
研究人员还进行了丰富的消融实验,揭示了不同模块设计对性能的影响,相关模型细节已在NeurIPS 2024的xTrimoGene模型中发表。

综上所述,scFoundation模型为建立细胞预训练大模型的模型架构、训练框架,和下游示范应用体系都提供了新的思路和方法,为生物医学任务的学习提供了基础功能,拓展了单细胞领域基础模型的边界。
目前模型权重及代码已开源。同时为了减少计算负担,支持更多用户轻量使用,研究团队也提供了模型相应的API,用户可在线获取scFoundation模型表征,支持CLI、Python SDK和网页端调用。
相关推荐




- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。










