

 新火种
2023-12-21
新火种
2023-12-21 


人工智能为新药铺平道路:几何深度学习方法可以预测合成药物分子的最佳方案

编辑 | 萝卜皮
后期功能化是优化候选药物特性的一种经济方法。然而,药物分子的化学复杂性往往使得后期多样化具有挑战性。
为了解决这个问题,德国慕尼黑大学(Ludwig-Maximilians-Universität München)、苏黎世联邦理工学院(ETH Zurich)和巴塞尔罗氏制药(Roche Innovation Center Basel)的研究人员开发了基于几何深度学习和高通量反应筛选的后期功能化平台。
考虑到硼基化是后期功能化的关键步骤,计算模型预测了不同反应条件下的反应产率,平均绝对误差范围为 4-5%;模型对已知和未知底物的新反应的反应性进行分类,平衡准确度分别为 92% 和 67%。主要产物的区域选择性被准确捕获,分类器 F 分数为 67%。当应用于 23 种不同的商业药物分子时,该平台成功发现了许多结构多样化的机会。
该研究以「Enabling late-stage drug diversification by high-throughput experimentation with geometric deep learning」为题,于 2023 年 11 月 23 日发布在《Nature Chemistry》。

LSF项目在药物化学研究中有着举足轻重的作用
当旨在建立药物化学中的结构-活性关系时,结构的新颖性和复杂性使得化学目标结构的合成具有挑战性。构效关系模型指导先导化合物和先导化合物优化方案,旨在提高候选药物的药理活性和理化性质。对于结构-活性关系探索,高效的整合非常重要,这是设计-制造-测试-分析周期的瓶颈。
许多选择性激活和修饰 C-H 键的合成方法可实现有机支架的后期功能化 (LSF),范围从分子构件到高级药物分子。许多催化系统提供定向和非定向方法,以及对修饰类似物的化学和位点选择性访问。
在众多 LSF 方法中,C-H 硼化被认为是最通用的快速化合物多样化方法。有机硼物质可以转化为一系列官能团,并作为后续 C-C 键偶联的稳健手柄,从而实现广泛的结构-活性关系研究。
然而,迄今为止,LSF 在药物发现中的应用仅有少数报道。大多数这些罕见的例子都集中在单一的 LSF 反应类型上。多个官能团和具有不同键强度、电子特性以及空间和官能团环境的各种类型的 C-H 键对直接 LSF 提出了挑战。并且开展 LSF 项目通常非常耗时且需要资源密集的实验,这与许多药物化学项目的紧迫时间表和有限资产不相容。
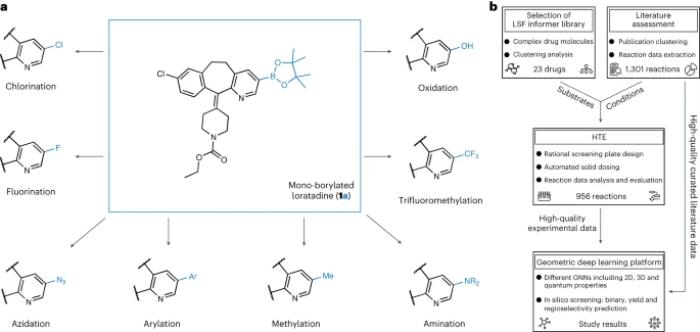
图示:硼化多样化研究概述。(来源:论文)
人工智能支持的 LSF
高通量实验 (HTE) 是一种既定的反应优化方法,可实现半自动小型化小批量筛选,从而快速、可重复地使用少量珍贵的构建模块和耗材并行执行多个转化。结合可生成有关成功和失败反应的高质量数据集的 FAIR(可查找性、可访问性、互操作性、可重用性)文档,HTE 通过实现高级数据分析和机器学习,为解锁 LSF 进行药物发现奠定了基础。
图神经网络(GNN)在分子特征提取和属性预测方面有着广泛的应用。在为化学反应规划开发的各种机器学习方法中,GNN 已成功应用于逆合成规划、区域选择性预测和反应产物预测。此外,还开发了 transformer 和基于指纹的方法来解决类似的问题。
有研究表明,学习过渡态几何结构的活化能可以准确预测竞争反应结果。使用密度泛函理论(DFT)级原子部分电荷的图形特征化改进了对电子效应驱动的反应的区域选择性的预测。图机器学习与 HTE 的结合可以优化有机底物 C-H 活化的反应条件。有些研究重点关注使用过渡态的深度学习模型,这类模型具有预测反应结果的能力,包括在某些情况下的对映选择性。
然而,这些方法仅限于小分子结构和相对较小的数据集,使得将此类模型应用于结构更复杂的药物样分子具有挑战性。文献里的研究表明,通过过渡态的量子化学信息增强的混合机器学习模型,能够对铱催化的硼化反应进行区域选择性预测。但是,空间效应和电子效应对 C-H 活化反应模型性能的影响,及其在具有多个芳环系统的分子区域选择性中的应用,仍未得到探索。
自动 LSF 硼化筛选的几何深度学习
慕尼黑大学、苏黎世联邦理工学院和巴塞尔罗氏制药的研究人员介绍了一种应用于自动 LSF 硼化筛选的几何深度学习方法,用于识别后期命中和先导多样化机会。采用计算深度学习来预测复杂药物分子 LSF 的反应结果、产量和区域选择性。
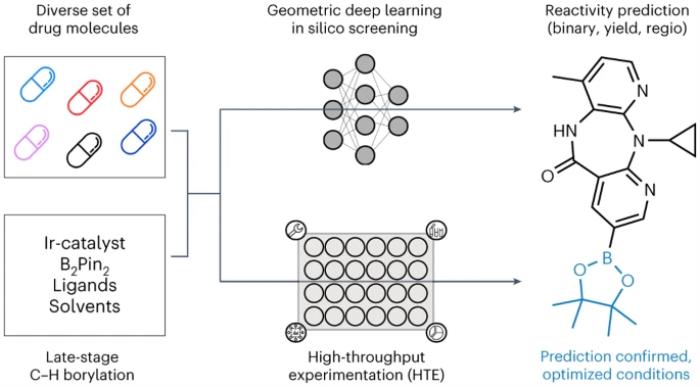
「这种方法有可能显著减少所需的实验室实验数量,从而提高化学合成的效率和可持续性。」论文的主要作者,慕尼黑大学化学与药学学院和罗氏公司 David Konrad 博士研究小组的博士生 David Nippa 说。
在该研究的第一步中,对已发表的文献进行了全面分析,为选择合适的 HTE 筛选反应条件和反映药物发现后期先导化合物性质的相关底物提供了理论依据。反应条件是根据 38 篇选定文献数据集手动整理的文献数据选择的。
LSF 底物的选择基于对 1,174 种已批准药物的聚类分析,产生了 23 种结构不同的药物分子。这种方法使研究人员能够在「信息库」方法(即涉及为评估合成方法而定制的化学空间的方法)中使用反应条件和底物的相关示例,而不是使用适用性有限的理想化底物和片段来优化先导化合物。
在第二步中,研究人员使用半自动化 HTE 生成数据(实验数据集)。所选药物分子和反应条件的反应数据为后续反应结果的机器学习提供了高质量的数据。
最后,不同的 GNN 在二维、三维和原子部分电荷增强分子图上进行训练,从而预测二元(是/否)反应结果、反应产率和区域选择性。「有趣的是,当考虑起始材料的三维信息而不仅仅是其二维化学式时,预测结果会得到改善。」苏黎世联邦理工学院的博士生 Kenneth Atz 说。
该方法已成功用于识别现有活性成分中可以引入额外活性基团的位置。这有助于研究人员更快地开发已知药物活性成分的新的、更有效的变体。
相关推荐




- 免责声明
- 本文所包含的观点仅代表作者个人看法,不代表新火种的观点。在新火种上获取的所有信息均不应被视为投资建议。新火种对本文可能提及或链接的任何项目不表示认可。 交易和投资涉及高风险,读者在采取与本文内容相关的任何行动之前,请务必进行充分的尽职调查。最终的决策应该基于您自己的独立判断。新火种不对因依赖本文观点而产生的任何金钱损失负任何责任。










